The Ultimate Guide to QMS Software
Q uality is always a top priority for life sciences companies looking to develop safe and effective products. To maintain a competitive advantage in today’s global market, companies are tasked with maximizing productivity, while meeting customer expectations and regulatory requirements. To meet these goals, many organizations rely on enterprise quality management system (eQMS) software solutions to get high-quality products to market fast. In this guide, we reveal everything you need to know about QMS software and outline the key factors that you should consider when exploring the best way to establish compliant quality management processes.
uality is always a top priority for life sciences companies looking to develop safe and effective products. To maintain a competitive advantage in today’s global market, companies are tasked with maximizing productivity, while meeting customer expectations and regulatory requirements. To meet these goals, many organizations rely on enterprise quality management system (eQMS) software solutions to get high-quality products to market fast. In this guide, we reveal everything you need to know about QMS software and outline the key factors that you should consider when exploring the best way to establish compliant quality management processes.
Why Is QMS Important?
Quality management systems help drive continuous improvement and provide a systematic approach to identify quality issues, improve product performance, lower manufacturing costs, and reduce waste. More and more organizations rely on a QMS to comply with FDA, EU MDR, ISO, and other regulatory standards, thereby reducing audit risks and consequences.
What Is QMS Software?
Traditional paper-based approaches to managing quality records make it difficult for remote teams to find files and identify the latest revisions since they are typically stored in multiple physical locations. It is also challenging for staff to track corrective and preventive actions (CAPAs) and demonstrate closed-loop quality processes. Moreover, getting documents through change cycles can be time-consuming and prone to errors with manual, paper-driven processes.
QMS software addresses these obstacles by providing organizations a central repository to document, store, and maintain quality records. Because records are managed electronically, it is easier to establish revision controls and audit trails. Eliminating paper also reduces the likelihood of errors due to manual changes, lost documentation, and misinterpretation of information.
Modern product-centric QMS software solutions enhance quality management even further by enabling organizations to link quality information such as CAPAs, training records, and design history files (DHFs) to the product record in one shared system. Engineering, quality, manufacturing, related product teams, and supply chain partners can collaborate in real time on the latest document changes, product changes, and issues and speed the review and approval process. Because a complete history is readily accessible, teams gain greater visibility and traceability over quality processes and reduce compliance risks.
Cloud-native QMS solutions sold under software-as-a-service (SaaS) models eliminate collaboration latency, data errors, and security issues with a persistent digital thread that leads to greater audit confidence and faster product launches. By utilizing a pay-as-you-go subscription, SaaS QMS software also reduces infrastructure expenses and large upfront software purchases that are associated with traditional on-premises platforms.
8 Essential Elements of Enterprise QMS (eQMS) Software
- BOM Management
The ability to effectively manage your complete product record, including the bill of materials (BOM), ensures that the most current product revisions are always controlled and accessible by all members of the quality, product, and extended supply chain teams. It also ensures that device master records (DMRs), design history files (DHFs), parts, standard operating procedures (SOPs), and drawings are connected to the product record making it easier to manage closed-loop quality processes. - Engineering Change Management
Engineering change management provides controlled design change processes that speed product development. Product team members can process formal engineering change requests (ECRs) and engineering change orders (ECOs) more efficiently with automated approval routings. Team members can also document the full history of product changes, which is critical to address internal or external audits. - Regulatory Compliance
To mitigate compliance risks, QMS software should help you adhere to FDA, ISO, and other key industry regulations or standards. It should enable the implementation of closed-loop corrective and preventive action (CAPA) processes to speed resolution of issues and prevent future occurrences of nonconformance. QMS should also provide complete control and traceability of product and quality records including DMRs, DHFs, SOPs, and training records. - Design Controls
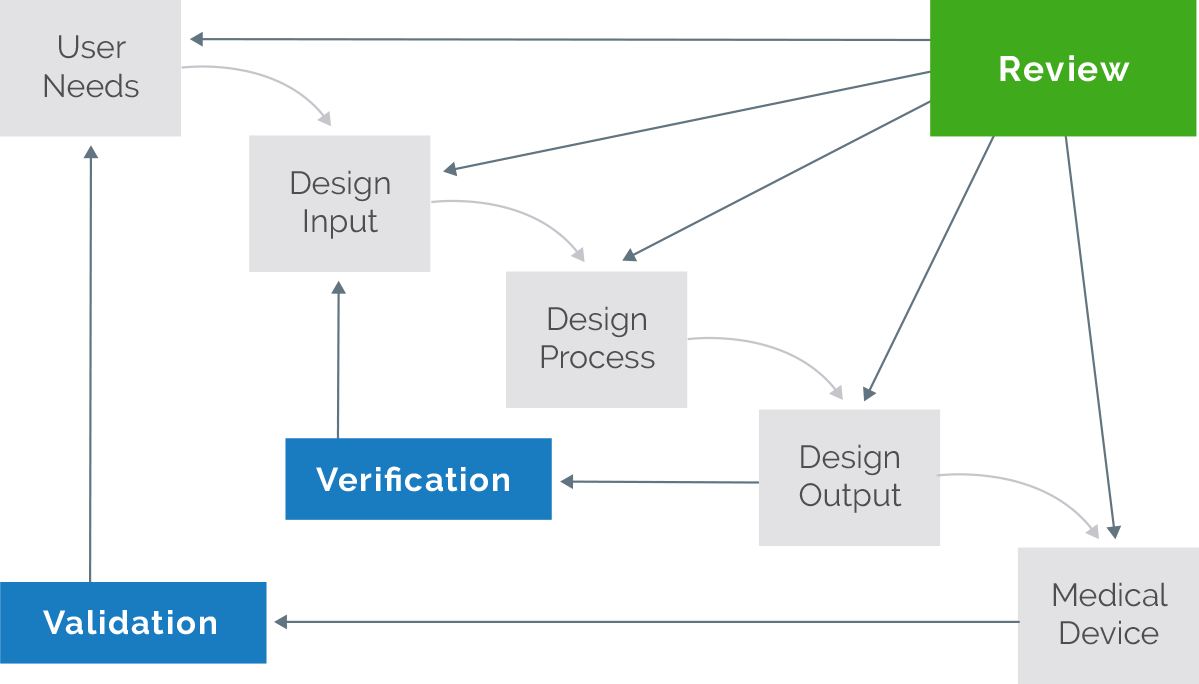
 Effective QMS solutions should establish and track design control elements such as user needs, design inputs, design processes, and design outputs. This helps simplify your review, verification, and validation processes. More importantly, a QMS should help you demonstrate compliance with policies, procedures, and FDA Title 21 CFR Part 820 as you move from early design phases through commercialization.
Effective QMS solutions should establish and track design control elements such as user needs, design inputs, design processes, and design outputs. This helps simplify your review, verification, and validation processes. More importantly, a QMS should help you demonstrate compliance with policies, procedures, and FDA Title 21 CFR Part 820 as you move from early design phases through commercialization. - Requirements Management
Requirements management helps teams ensure that their product development objectives are met during a product lifecycle. The ability to link requirements to the product record enables organizations to manage and track issues, defects, and customer needs throughout the entire lifecycle and innovate more rapidly. Effective requirements management processes create tighter connections and better collaboration between the project team members, managers, suppliers, and other key stakeholders. - Document Management
To meet the demands of highly regulated environments, companies must be able to store, track, and manage all of the documentation associated with product and quality records. This includes product specifications, assembly instructions, SOPs, quality policies, and training records. Document management allows for the centralized storage of documentation, as well as the establishment of audit trails and revision control. - Training Management
Product companies must maintain an employee training program to demonstrate compliance with FDA, ISO, safety, internal procedures, and other regulatory standards. Having an efficient, automated system to create employee training plans, assign training records, and generate progress reports helps simplify audits and reduce compliance risks. - Product-Centric FrameworkThe key to introducing high-quality products is maintaining a tight connection between all of your product information, quality records, training plans, and compliance processes.
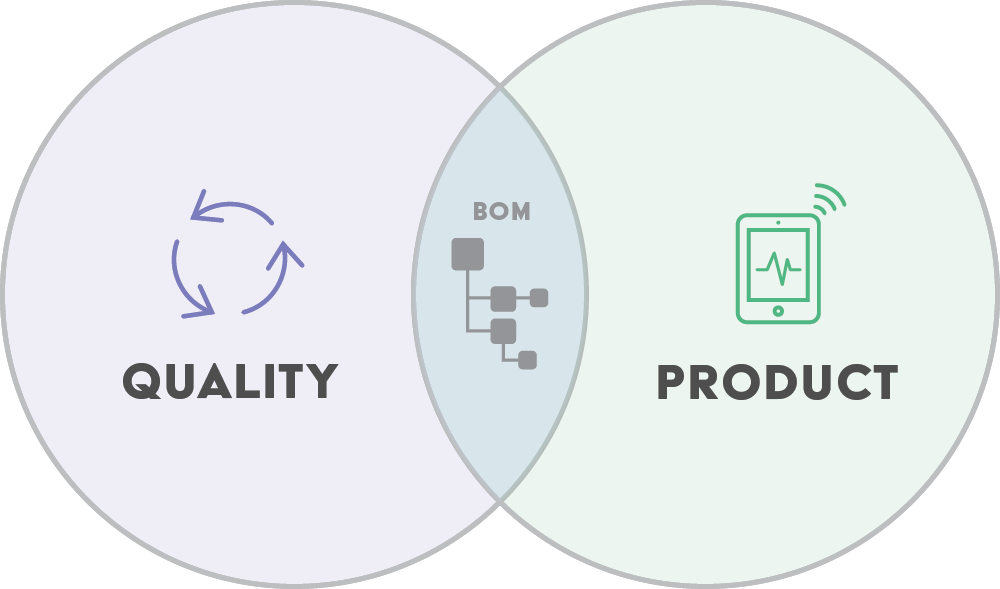
Product-centric QMS platforms manage linked relationships between DMRs, DHFs, BOMs, individual components, approved manufacturer/supplier lists (AMLs/ASLs), documentation, product history, and any changes or quality issues. This enables full visibility and traceability throughout new product development and introduction and ensures regulatory compliance.
Things to Consider When Implementing a QMS SOFTWARE SOLUTION
Consider these key questions when evaluating what it takes to implement a QMS software solution successfully.
- Software Validation
Does the QMS software vendor provide a software validation service to help you meet the installation qualification (IQ) and operational qualification (OQ) requirements set forth by the FDA? Will the vendor provide the necessary IQ and OQ documentation set with every software release to support an FDA audit or submittal? - Security
Is the QMS solution designed with a multilayered security model to ensure that your intellectual property (IP) is always protected? Are there safeguards or access controls in place to further protect product information and privacy? - Disaster Recovery
Is the QMS software designed with built-in redundancies to quickly recover from a wide range of system failures? - Implementation and Training
What resources are required from the QMS vendor and your team to complete the software implementation? How much time is needed to configure, test, and go live? What is the QMS vendor’s approach to training? Is it a train-the-trainer approach, or will your key stakeholders need to participate in the initial onboarding process? Is training provided via live or recorded sessions? - Ongoing Customer Education and Support
Do you have access to adequate support and educational resources (e.g., best-practice information) after go-live? Are live, dedicated support staff (e.g., customer success coaches, customer service representatives) available to answer your questions to help drive your successful adoption and expansion of the QMS platform? - Scalability
Is the QMS platform designed to easily accommodate additional users, suppliers, and/or functionality as your company continues to grow and product processes evolve? - Total Cost of Ownership (TCO)
Your TCO should include not only up-front software costs—but costs associated with ongoing maintenance. Determine if there are additional costs for annual maintenance and/or new software releases. If so, what is the estimated frequency of upgrades? Also identify any costs associated with training and customer support. SaaS platforms include customer support as part of the software subscription and there are no additional charges to take advantage of regular software enhancements.
QMS Resources
ebooks
7 Principles of Product-Centric Quality Management
Audit Resilience in a Virtual World: A Practical Guide
White Papers
How Strong Design Controls Simplify Compliance and Eliminate Audit Anxiety
FDA Software Validation: How Cloud QMS Reduces Costs and Resource Drains
Meeting Regulatory Compliance Requirements in Today’s Global Market
Best Practices
How to Classify Your Medical Device for FDA Approval
5 Tips to Pass Your FDA or ISO Audit with Confidence
The Cost Savings Calculator