What Are the Key Elements of a Quality Management System (QMS)?
 In today’s highly competitive landscape, life sciences companies are tasked with getting quality products to market faster while meeting customer expectations and regulatory requirements. Having an effective quality management system (QMS) in place is critical to help organizations reach their product development milestones and achieve commercialization success.
In today’s highly competitive landscape, life sciences companies are tasked with getting quality products to market faster while meeting customer expectations and regulatory requirements. Having an effective quality management system (QMS) in place is critical to help organizations reach their product development milestones and achieve commercialization success.
QUALITY MANAGEMENT SYSTEM OVERVIEW
A QMS is a formalized business system of documenting policies, processes, responsibilities, and procedures to meet customer requirements as well as compliance with FDA, ISO, CE, UL, and other regulatory standards. An effective QMS enables regulated companies to sell and sustain safe and effective products that consistently meet or exceed customer expectations.
The implementation of a QMS typically starts at the executive level, where business goals and objectives are defined. These goals and objectives translate into policies, processes, and standard operating procedures (SOPs) that are adopted across the entire organization.
What Is the Purpose of a QMS?
QMS provides the framework to ensure that processes are carried out in a systematic manner. It also allows for continuous improvement. Organizations use QMS as a roadmap for meeting regulatory requirements and achieving their customer satisfaction and revenue goals.
COMMON QUALITY SYSTEM STANDARDS
ISO 9001 Compliance
ISO 9001:2015 is a standard within the ISO 9000 family of international quality standards that is considered to be the most widely adopted approach to quality management systems. Many organizations use ISO 9001 as the basis to develop their own QMS program. Other ISO standards related to QMS include:
- ISO 13485 (QMS for medical devices)
- ISO 14971 (Risk management for medical devices)
- ISO 9004 (Continuous improvement/guidance to achieving sustained success)
U.S. Food and Drug Administration (FDA) Compliance
The FDA mandates that medical device, biotechnology, and other life sciences companies must establish and follow quality systems to ensure their products consistently meet applicable requirements and specifications for the U.S. market. While quality management system definitions vary, the FDA’s quality system regulation specified under Title 21 CFR Part 820 establishes that medical device manufacturers must establish and follow current good manufacturing practices (CGMPs) for the design, manufacture, installation, and servicing of safe and effective devices1. In the near future, the FDA is expected to revise its current Title 21 CFR Part 820 quality system regulation to more closely align with ISO 13485. This will enable medical device manufacturers to have a more globally harmonized QMS.
Design controls are at the heart of the FDA’s quality system regulation and ISO 13485. The FDA specifies the necessary steps and procedures for each design phase. These design controls govern the processes throughout new product development and introduction (NPDI). The FDA also defines how manufacturers demonstrate compliance. This includes the provision of two pieces of compliance evidence—the design history file (DHF) and the device master record (DMR).
Medical device companies that use electronic systems to track DMRs, employee training records, corrective actions, or other data requiring traceable documentation as part of their QMS must also comply with FDA Title 21 CFR Part 11. This regulation establishes the criteria under which electronic records and signatures are deemed trustworthy, reliable, and generally equivalent to those executed on paper.
EU MDR Compliance
Regulatory standards for medical devices in the European Union (EU) are established through the EU Medical Device Regulation 2017/745 (EU MDR) by the European Commission. Medical devices and other products covered under the EU MDR are required to have a Conformitè Europëenne (CE) Mark in order to be sold in the European Economic Area (EEA). The CE marking (symbol) represents a manufacturer’s declaration that products comply with all applicable European laws or directives with respect to safety, health, environment, and consumer protection. Medical device manufacturers must select an appropriate route for conformity assessment (i.e., Annex IX, X, or XI). The required route is determined by the classification of the medical device.
Conformity assessment routes:
- Annex IX (QMS and technical documentation) is used when a full QMS is implemented by the manufacturer. A review of technical documentation is also required with or without the issuance of certificate.
- Annex X (type-examination) is used when a manufacturer wants to certify a device based on a representative sample. A notified body examines and/or tests the representative sample and associated technical documentation to determine if the device meets MDR requirements.
- Annex XI (product conformity verification) is used in association with Annex X or in combination with technical documentation for low-risk devices.
Because the conformity assessment routes include design verification and validation review, having an existing QMS that supports ISO 13485 compliance can help medical device manufacturers readily meet the necessary requirements of the EU MDR.
Medical Device Classifications
The regulations that apply to a particular medical device vary based on device classifications. The FDA and European Commission use different criteria for classifying medical devices. The classifications are typically related to the product’s intended use, indications for use, and the risk that it poses. Intended use describes the general purpose or function of the medical device, whereas indications for use describe the disease or condition the medical device will diagnose, treat, prevent, or cure. They also describe the target patient population.
The FDA categorizes medical devices as either Class I, Class II, or Class III. Different regulatory controls (i.e., General, Special, or Premarket Approval (PMA)) are assigned to each classification to provide reasonable assurance of the safety and effectiveness of the device. The amount of regulatory control increases as the device progresses from Class I to Class III. General controls apply to all medical devices unless exempted by regulations. Special controls are regulatory requirements that apply to Class II devices. These controls are device-specific and include performance standards (i.e., design characteristics or specifications), postmarket surveillance, patient registries, special labeling requirements, premarket data requirements, and guidance documents. Premarket Approval (PMA) is required for Class III devices. As part of the PMA application, extensive scientific evidence in the form of laboratory and clinical trial data must be submitted to the FDA to prove the safety and effectiveness of the device.
Devices that do not require PMA will most likely require a 510(k) premarket notification to obtain FDA clearance. The 510(k) is a faster and less expensive submission route that can be used if the device is substantially equivalent to a device that is already cleared by the FDA. A 510(k) submission typically includes detailed technical, safety, and performance information about the medical device.
- Class I devices pose a low to moderate risk to patients and/or intended users. Today almost 50% of medical devices fall into the Class I category. Devices in this category are subject to general regulatory controls; however, a majority are exempt from 510(k) premarket notification, and FDA clearance is not required before selling these devices in the United States. Examples of Class I devices include bandages, disposable gloves, tongue depressors, medical thermometers, and stethoscopes.
- Class II devices pose a moderate to high risk to patients and/or intended users. Today over 40% of medical devices fall into the Class II category. Devices in this category are subject to both general and special regulatory controls, and a majority require a 510(k) premarket notification to obtain FDA clearance. Examples of Class II devices include catheters, syringes, contact lenses, and pregnancy test kits.
- Class III devices pose a high risk to patients and/or intended users. Only 10% of medical devices fit into this category. Class III devices are subject to an application for PMA which requires extensive clinical data to support their safety and effectiveness. These devices typically help sustain or support life and can also be implanted. Examples of Class III devices include defibrillators, pacemakers, breast implants, and implanted prosthetics.
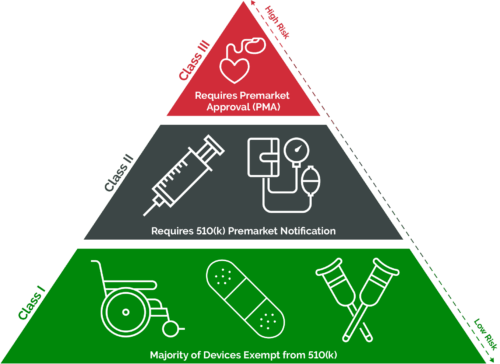
FDA requirements for Class I, II, and III devices are further defined in Title 21 CFR Part 820 and ISO 13485.
Classification of medical devices in the EU is regulated by Annex VIII of the Medical Device Regulation (MDR). Similar to the U.S., devices are classified as:
- Class I device (low risk)
- Class IIa device (low to moderate risk)
- Class IIb device (moderate to high risk)
- Class III device (high risk)
Under the EU MDR, the classifications above are based upon whether a device is deemed noninvasive, invasive, or active as well as its duration of use (i.e., transient, short-term, or long-term). The classification of the medical device will determine the route in which a device obtains its CE Mark (i.e., Annex IX, X, or XI).
EU requirements for Class I, IIa, IIb, and III devices are further defined in EU MDR 2017/745.
Underwriters Laboratories
Agencies like Underwriters Laboratories (UL) set industry-wide safety and quality standards for new products. UL testing makes sure that electric circuitry is correct and that devices can handle the amount of current as claimed by the manufacturer. They also ensure that products are constructed correctly for the highest safety. Products that are UL tested typically carry a UL stamp (similar to the CE Mark) and are assigned one of the following UL designations:
- UL Listed: This designation means that the product meets the laboratory’s standards and can be used by itself for a specific purpose.
- UL Recognized: This designation is granted to equipment components that are used in combination with other pieces of equipment to create a finished product.
- UL Classified: This designation means that a product has only been evaluated for specific properties regarding a limited range of standards or for use under certain conditions.
8 KEY ELEMENTS OF AN EFFECTIVE QMS
When setting out to implement a new quality management system or fine-tune an existing one, organizations should keep these key elements in mind.
1. Quality Policy and Objectives
A quality policy is a brief statement that outlines an organization’s overall purpose and mission and supports its strategic direction. It also states the company’s commitment to continuous improvement as well as meeting all applicable regulatory and customer requirements. A quality policy serves as the framework for the entire QMS.
Quality objectives are measurable goals that stem directly from the quality policy. Quality objectives should contribute to the enhancement of customer satisfaction and are commonly tied to areas such as:
- Product nonconformities or defects
- Product performance
- Efficiency
- Safety
- Delivery
- Customer service
For each of these areas, establish quantifiable targets and link to key performance indicators (KPIs) that help monitor the achievement of the objectives over time. It is best practice to make quality objectives SMART: Specific, Measurable, Achievable, Relevant, and Time-bound.
Here is an example of a quality objective that is tied to customer service:
- Objective: Reduce customer complaints
- Target: Achieve 25% reduction within the next three months
- KPIs: Customer call reports or customer satisfaction surveys
The setting of objectives and targets involves individuals from key functional areas across the organization. Once established, quality objectives should be clearly documented and communicated throughout the organization. It is also important to update quality objectives as business needs continue to evolve.
2. Quality Manual
A quality manual documents the scope of the QMS. The manual includes information about the organization’s objectives, expectations, policies, processes, and more. The manual also includes requirements needed for an organization to comply with ISO, FDA, and other regulatory standards.
3. Organizational Structure and Responsibilities
A quality management system outlines the roles and responsibilities of the key stakeholders, resources, and infrastructure that are needed to achieve an organization’s quality objectives. It also provides a clear visual of how the organization is structured. This can be in the form of a flow chart or a similar type of diagram.
4. Internal Processes
An organization should identify and define processes that require any type of resource to transform inputs into outputs. Resources can encompass employees, machines, or technology. Establish and document standards and metrics for measuring the performance of these processes to ensure that quality outputs are always the end result.
5. Customer Satisfaction With Product Quality
As part of QMS, organizations are required to monitor customer satisfaction to determine if their quality objectives are being met. This can be in the form of customer satisfaction surveys, customer complaint reports, customer review websites, and other monitoring systems.
6. Continuous Improvement
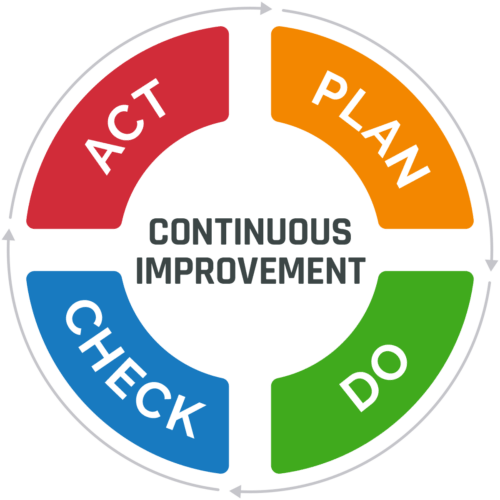
Continuous improvement is the ongoing improvement of products, services, or processes through incremental changes over time or changes that occur all at once. A widely used tool for continuous improvement is a four-step quality assurance method called the Plan-Do-Check-Act (PDCA) cycle:
- Plan: Identify an opportunity for improvement and plan for change
- Do: Implement the change on a small scale
- Check: Use data to analyze the results of the change
- Act: If the change was successful, implement it on a wider scale and continuously assess the results. If the change does not work, begin the cycle again.
Other common tools include Six Sigma, Lean, and total quality management. These approaches emphasize employee engagement and teamwork, as well as practices to reduce variation, defects, waste, and cycle time.
7. Document Control
Organizations should have a system in place for document control. This includes:
- Review and approval of documents for adequacy prior to release
- Review/update/re-approval of documents on an as-needed basis
- Identification of changes and current document revision status
- Preventing obsolete documents from unintended use
- Policy or procedure for the identification, storage, protection, retrieval, retention, and disposal of documents
8. Enterprise Quality Management System (eQMS) Software
Once you have established your quality policy and objectives and other key processes for your quality management system, how do you ensure that it yields the desired results?
Today, more and more businesses are relying on some type of eQMS software to centralize all of the key elements of their quality and product records and allow for more effective collaboration across dispersed teams and partners.
Cloud-native QMS platforms help eliminate nonconformance issues, reduce audit risks, and improve new product development processes by improving quality controls and documenting policies, procedures, and practices..
How To Buy The Right Software

Choosing the right software for your business needs can be challenging.
Our step-by-step Software Buyer’s Guide walks you through how to evaluate and select the right cloud software solution to meet your specific requirements. We share the collective wisdom of many leaders’ experiences and recommend best practices to help you avoid common mistakes. You’ll learn how to identify requirements, compare vendor solutions objectively, and make compelling software purchasing recommendations to your executive team.
So, don’t reinvent the wheel and waste countless hours and resources. Read our definitive guide to learn how to make your next software-buying decision a snap.
ADOPTING A PROUCT-CENTRIC APPROACH TO QUALITY MANAGEMENT
While traditional document-centric QMS solutions simply automate paper-driven processes and provide basic documentation control for standard operating procedures (SOPs) and other files, product-centric QMS software maintains the complete product record and associated quality processes in a single platform. By managing dynamic relationships between the product bill of materials (BOM), CAPA processes, design history files (DHFs), engineering changes, and other critical records, teams gain greater traceability and visibility throughout the entire product development cycle to avoid compliance risks and bring compliant products to market fast.
QMS Resources
eBooks
- 7 Principles of Product-Centric Quality Management
- Audit Resilience in a Virtual World: A Practical Guide
- How to Buy the Right Software
White Papers
- How Strong Design Controls Simplify Compliance and Eliminate Audit Anxiety
- FDA Software Validation: How Cloud QMS Reduces Costs and Resource Drains
- Meeting Regulatory Compliance Requirements in Today’s Global Market
- The Essential Guide to EU MDR Compliance
- The Essential Guide to EU IVDR Compliance
Ready to Learn More?
Four Companies With Exceptional Quality Management System
Ultimate Guide to QMS Software
References