Managing The Device Master Record (DMR) to Comply with 21 CFR Part 11 and Part 820
What is the Device Master Record
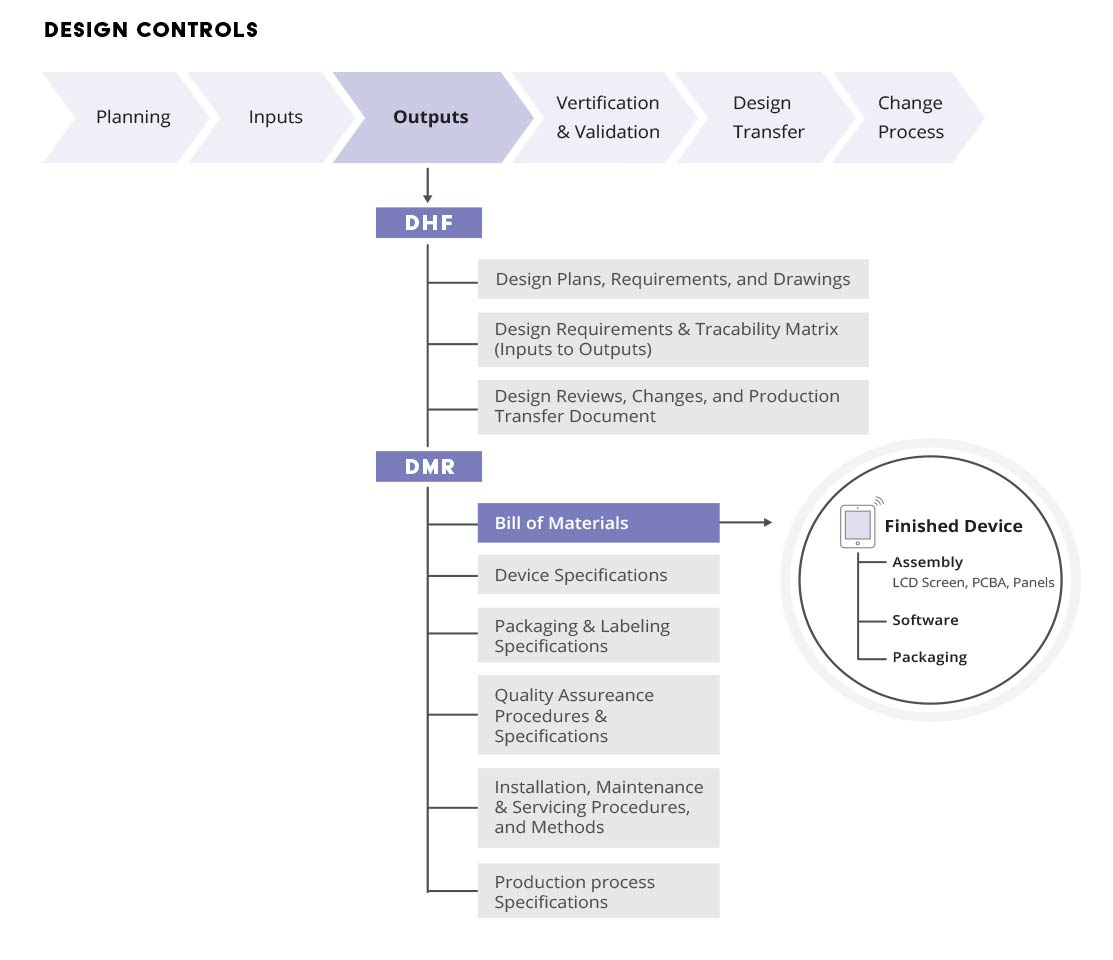
The Device Master Record (DMR) is an all-encompassing collection of documents and records (including device specifications, process specifications, quality assurance procedures, and packaging and labeling specifications) mandated by 21 CFR Part 820.181.
Developing medical devices requires compliance with the FDA, ISO, EMA, and other regulatory bodies. Strong design controls simplify your review, verification, and validation processes. Furthermore, it speeds design changes and transfers by establishing and tracking design control elements: user needs, design inputs, design processes, design outputs, and device documentation.
Document control in medical device companies
In the world of medical device development and manufacturing, document control is critical to company success. Engineering records drive medical product development, and the company quality system and Standard Operating Procedures (SOPs) govern how business is conducted. Organized, revision-controlled, and auditable records are fundamental to maintaining regulatory compliance.
Traditionally, companies have created and updated company data in design programs, SOPs, memos, and spreadsheets. Changes are printed out and routed for signatures in colored folders. Product information is communicated to internal teams, outsourced manufacturers, and suppliers via email, phone, or disconnected chat tools. Critical product records, including the design history file (DHF), device master record (DMR), and bill of materials (BOM) are kept in a combination of engineering vaults, spreadsheets, and paper files. Final BOMs are usually loaded into enterprise resource planning (ERP) systems.
However, compressed product lifecycles, stringent regulatory requirements, geographically dispersed project teams, cost pressures, and outsourced partnerships are challenging traditional document control and communication in a number of ways:
- Modern product records (e.g. DHFs, DMRs, BOMs) are large, complex, and continually evolving. Manually making and controlling changes in distributed locations and spreadsheet formats is inefficient and error-prone, and makes maintaining FDA-required traceability difficult.
- Records are highly relational and include various associated data and files. Within the device master record (DMR) and design history file (DHF) for each product, all revisions, parts, and drawings must remain properly linked and tracked. Compliance data should be attached to item records. Spreadsheets, servers, and paper files alone provide no integrated way to aggregate all related item data.
- Maintaining a system of spreadsheets and servers in a validated state is highly challenging. Device master record (DMR) and design history file (DHF) records must be available for quality audit by the FDA at any time, and backed up and retained for a minimum of 2 years. The fluid nature of spreadsheets does not lend itself to the level of organization and accountability required in the medical device industry.
- Spreadsheets and ERP systems do not have integral processes for managing engineering change orders (ECOs) and cannot be used to control device master record (DMR) changes, manage associated files or create proper documentation for the DHF. FDA regulations require that ECOs be properly verified, validated, reviewed, and documented in the DHF of the product. Manual routing of paper ECOs in colored folders is inefficient and prone to bottlenecks.
- Throughout the lifecycle of a product, numerous revisions of product information are created and communicated to multiple internal and external teams. Keeping everyone on the latest revision of product data when it is spread over local computers and in various spreadsheets and paper files is a nearly impossible feat. It is especially difficult for external partners and suppliers to obtain current product information.
How automated solutions help maintain the DMR
Today automated solutions are available to help companies organize, maintain and communicate product information—including the device master record (DMR), design history file (DHF), and BOM—and succeed in a competitive and highly regulated environment. Companies accelerate their new product introductions, improve collaboration with outsourced partners and suppliers, and achieve compliance with confidence. In a study by Aberdeen Group, a global research consultancy, they reported that best-in-class companies significantly outperform laggards in new product development (Figure 1). These companies tend to leverage these tools to improve product development performance and profitability. They are over 50 percent more likely to “leverage centralized product repositories to better capture, share and reuse core information such as items and bills of materials (BOMs).”
As a provider of a collaborative BOM, device master record (DMR), and design history file (DHF) solution, Arena has worked with hundreds of companies across diverse industries to improve their product development and manufacturing processes. Arena Quality Management Systems (QMS) software is currently used by numerous medical device companies to manage their critical product data. Through years of working with medical customers, Arena understands the obstacles these companies face in effectively managing and sharing their product information. This white paper aims to provide insights into these challenges and discuss some innovative solutions designed to help medical device companies succeed.
Current product record maintenance and its challenges
Why spreadsheets and paper files are inadequate for managing DMR-related data
Throughout the design of a product, large amounts of critical data are created. A new medical device development effort begins with design and development planning and creation of initial quality documentation to govern the design process. Engineering groups then create designs using computer-aided design (CAD) tools and electronic design automation (EDA) applications. These designs originate as unreleased and flexible files. Part files, design data, and review information begin to aggregate as the basis of the device master record (DMR) and design history file (DHF). Since testing is integral to verification and validation efforts, the design then moves to initial test builds. Test data is created.
To communicate the component needs and cost estimates, an engineering project manager often creates an engineering BOM (EBOM) for the new product in a spreadsheet that also will become part of the device master record (DMR). A purchasing group may create an approved vendors list (AVL) or approved manufacturers list (AML). All of this unreleased data is typically corralled using a CAD vault system, a source code repository, individual file servers, spreadsheets, and paper files. The data is typically widely dispersed, and not managed by any one party.
As the design progresses toward production, data must be centralized and tightly revision-controlled. The design must be properly tested, verified, and validated, and product records officially released. During this centralization process, numerous project teams contribute to the official product data (Figure 2), and all contributions must be captured, approved, and documented.
The resulting product record, including the device master record (DMR) and design history file (DHF), is highly relational and includes various associated data and files, such as design drawings, software files, item files, costing information, compliance status, specification data, and sign-off information. Product records are also shared with quality and compliance teams, and with purchasing and supply chain management for collaboration and review.
In many cases, medical device companies use a system of servers, paper files, and spreadsheets to aggregate, share and control this data. Change management may be done manually using colored folders and manual signatures. In order to maintain the document control necessary to meet FDA regulations, it is often necessary to have one or more document control specialists involved in maintaining and organizing the product record information.
Figure 2 — Multiple teams contribute to record creation and changes
In the production phase, data control becomes even more critical and data dissemination even more widespread. At this point, product changes must move through a strict change control process and compliance concerns are paramount. Manufacturers, engineers, and management must all be in sync. Changes must be communicated to operations, manufacturing, and finance groups, who are typically using enterprise resource planning (ERP) systems to track materials planning, sourcing, and other production-related information after releasing to production.
ERP systems are not designed to be change control or file management tools and must be manually updated to reflect approved product changes. To update and change product information across electrical and mechanical CAD tools and ERP systems, many companies rely on spreadsheets to manage part changes, SOPs, and BOMs and to communicate them to project teams. Unfortunately, spreadsheets are not particularly well-suited for these jobs. DHF documentation necessary for FDA compliance must also be generated separately. This distributed document control process is inefficient and error-prone, at a time when time to market, compliance, and traceability are most critical. Makeshift document control tools do not provide the project collaboration capabilities necessary to manage product development tasks and milestones across a global supply chain.
Using spreadsheets and paper files for maintenance of the product record falls short in several areas—change and revision control processes, communication with project teams, and management of more than a single product.
Manual processes can make the device master record feel disorganized
A modern product record often includes a complex set of hundreds to thousands of structured items. Poring over thousands of rows and columns in a spreadsheet to modify data leads to errors. Rapid changes during the design and prototype phases result in a higher probability of making mistakes. Because a system of spreadsheets does not have integrated change management capability, changes are difficult to trace, appropriate compliance documentation is difficult to generate, and finding errors if they occur is very difficult.
Even after the first product is built, the product record will continue to evolve—due to bug fixes, design improvements, part substitutions, or supplier switches—until the product reaches its end of life. The time spent to manually make and document changes and fix mistakes throughout the lifecycle of a product may amount to a substantial delay in its shipment.
With multiple teams inputting frequent changes, manual revision control and documentation processes can easily become overwhelming and expensive. It is difficult to track where engineering change orders (ECOs) are in the change review process. Documentation specialists are required to spend large amounts of time filing product data, test information, and change information. In many cases, these document control specialists must actively mine data from the product development team and route change orders by hand using paper folders. The device master record (DMR) and BOM can feel chaotic and disorganized, or rigid and difficult for the engineering team to update. In either case, the change order process is inefficient and onerous. There is no “single version of the truth”—representing the latest product and change information—to which all project teams and management can consistently and confidently refer throughout the lifecycle of a product.
To attain FDA approval, it is critical that all product record changes are both documented and traceable, with appropriate sign-off by responsible parties. Revisions must be clearly verified and validated according to accepted processes and test results linked to design files in the DHF. It is nearly impossible to track these changes and their associated documentation with a collection of different spreadsheets, paper files, and manually routed folders.
Sharing product data like the DMR can be difficult
A wide variety of internal and external teams use product record information. However, companies typically do not want to share the entire device master record (DMR) with design partners or suppliers. As a result, companies often divide product data into subsets to be shared with different audiences. Controlling access to ensure that each audience receives only the information they need becomes difficult as the supply chain expands in depth and complexity.
Email or fax is often used to share spreadsheet-based product information (like the BOM, lists of changes, and testing data) with project teams in diverse geographic locations. Over time, different revisions of spreadsheets may scatter on various desktops in different organizations. And as a result, the sourcing team may order incorrect parts or the contract manufacturer (CM) may execute a wrong build. These mistakes lead to excess or obsolete inventory, as well as significant recall, scrap or rework, all of which directly impacts the bottom line. In a medical application, a build error could have life- and company-ending consequences.
Quality teams must also interact with project data as they prepare for FDA submissions and audits. These individuals are not always familiar with the engineering details of the product or design history. If the device master record (DMR) and design history file (DHF) are poorly organized, the job of the quality group can be very time-consuming and difficult. With scattered spreadsheets and dispersed product data, the quality team is often forced to scramble to meet deadlines and address audit questions. In some cases, FDA compliance can be in jeopardy.
Real-time access to accurate product information is a key success factor in medical product development, manufacturing, and compliance. Companies must ensure that engineering and operations teams, design partners, quality team members, outsourced manufacturers, and suppliers all have secure access to a single, unified version of product data. Spreadsheets and manual communication tools do not lend themselves to providing and sharing this single version of complete and traceable product information.
Managing beyond a single product
Many companies have multiple products with layers of data that are interrelated, such as part data, mechanical files, electrical component sheets, bills of materials, the approved vendor list, and cost data. In a manual spreadsheet environment, the associated data is updated and communicated separately for each product, yet has to be kept in sync. For instance, BOMs and an item master are created and updated separately.
The inability of a company to manage information across multiple products limits its opportunities for categorization and part re-use. And when a supplier obsoletes an item, the company must find and change the item across multiple products that may contain that item. In the case that engineering leverages a BOM from a similar earlier design as a starting point for new product development, a lack of visibility into supplier parts may result in the use of obsolete parts. The issue will only be identified later in the design phase, potentially resulting in the costly redesign and retesting efforts.
With a spreadsheet-based BOM management system, files and documentation directly related to a part or assembly cannot be easily attached or shared with project teams. Communicating and tracking updates and changes to the product documentation is even more cumbersome. Inadequate in managing a single product record, spreadsheets and paper files fare even worse when it comes to managing multiple products and DMR- and DHF-related data, files, and documentation.
Next-generation document control
Device master record (DMR), design history file (DHF) and product data can be managed with automated tools
Many seasoned medical product development and manufacturing teams recognize the significant scope of today’s document control requirements and the severe limitations of spreadsheets and paper files in meeting their objectives. They fully recognize the need for a next-generation solution to replace inadequate manual spreadsheet tools. Demonstrating strong market growth in the past few years, collaborative BOM, DMR, and DHF solutions are effective tools that bring together the design, compliance, and manufacturing worlds (Figure 3). Ideal for medical device companies maintaining the device master record (DMR), design history file (DHF), and change process, solutions like Arena QMS successfully address challenges resulting from spreadsheet-based manual document management and inadequate communication channels (Table 1).
Table 1: Document Control Challenges and How Arena QMS Helps Medical Device Companies Manage BOMs, DMRs, DHFs and change orders
Typical Document Control
|
Arena QMS
|
|---|---|
| Product changes documented in the DMR and BOM are managed manually resulting in errors and delays. | Changes to product information are controlled and the change process is automated, traceable, documented, and auditable. |
| Change orders are routed in a manual and time-consuming process and are documented in a system separate from the product information to which they are related. | Change orders are routed electronically and automatically attached to relevant items and evidence records. Change order cycle times are dramatically reduced. |
| Multiple BOMs and item records exist on various desktops, resulting in design, sourcing, and manufacturing errors. | A single version of the truth—the latest product information, including the BOM, device master record (DMR), design history file (DHF) and change order information and status is captured in a centralized environment. The accuracy and efficiency of product record maintenance are tremendously improved. |
| Quality systems require constant supervision and maintenance to maintain a validated state. Tools require separate validation and record organization to prepare for audits results in high overhead. | A software-as-a-service (SaaS) system allows validation of the tool itself to occur at the software provider, with little work required by the user company. Data is organized and accessible from the beginning, with audit trails built-in, allowing constant audit readiness. A validated state is constantly maintained with little overhead. |
| Product record information is sent to internal teams and external suppliers via email or chat tools, causing delays and preventing effective collaboration. | The latest product information and changes are selectively accessible by project teams, internal or external, anytime and anywhere. The security of data is increased, as suppliers are given access only to the parts and assemblies they work with. Real-time access to information encourages and enables participation from partners and suppliers and improves collaboration across the global supply chain. |
| Compliance requirements are managed by manually working through a large volume of data. | Regulatory, environmental, and industrial requirements are managed early at the part level, with associated evidence files. Audit trails are automatically established with item records and change records. |
| The inability to manage across multiple products limits categorization capabilities and part re-use opportunities. | Product data is managed in a relational database where BOM items are associated with the item master, costs, AVL, etc. Items, costing data, supplier information, and other product details are easily accessed, changed, and managed across multiple products. |
| Attaching files and documentation to items is cumbersome. | Files and documentation can be attached to any items within the product record. Changes to these documents are tracked and controlled, and these changes are documented and auditable. |