Three Quality Subsystem Improvements That Streamline FDA Compliance
Introduction

Maintaining readiness for FDA inspections can be arduous and time-consuming, and the need for preparedness is growing. While the FDA continues to issue more observations than Form 483s, the number of observations has been decreasing, while the count of Form 483s has been increasing. A renewed focus on the quality subsystems that are most frequently cited with issues in Form 483 inspections can streamline FDA compliance and inspection readiness.
Here, we overview the subsystems that comprise the Quality Subsystem Regulation (QSR) and identify ways to improve QMS, the subsystems that show up in most industry-wide FDA warnings and citations. Additionally, we examine features and functions to consider in your own quality management system software solution to ensure compliance across these key subsystems.
What are the subsystems that make up the Quality System Regulation?
The FDA requires medical device manufacturers to establish a quality system that’s appropriate for the level of risk presented by a device, as well as the size and complexity of the activities and company that makes the device. The FDA’s direction is a rather loose framework that leaves a lot of room for manufacturers to build a system that meets their needs—or get paralyzed by the endless possibilities.
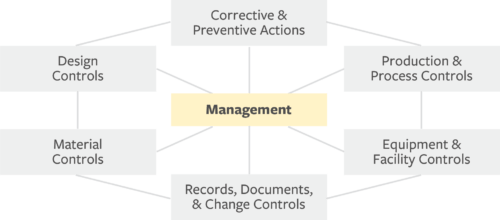
To provide guidance on how to build a quality system that meets the requirements of the QSR, the FDA outlines seven subsystems that should be built, documented, and implemented to support the development of safe and reliable products.
- Management: Lead design and execution of product design and quality systems
- Corrective & Preventive Actions (CAPA): Resolve and prevent quality problems
- Production & Process Controls (P&PC): Ensure device aligns with specifications
- Equipment & Facility Controls: Protect the design from negative environmental impact
- Records, Documents, & Change Controls: Facilitate and implement product and artifact changes for accurate, enduring design control record
- Material Controls: Maintain system to track and approve all materials and associated suppliers used in production
- Design Controls: Verify and validate design to user need and intended us
Start with these three subsystems
While efforts to improve performance across every quality subsystem are merited, it’s important for organizations to understand where focused efforts will yield the most gains. Larger, more complex manufacturers producing Class III medical devices will experience different issues than smaller manufacturers of simpler Class I medical devices. Product and company specifics warrant appropriate points of emphasis when driving improvement efforts.
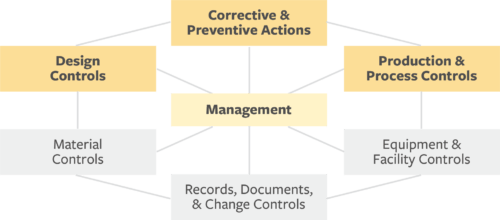
Regardless of your company’s product and organizational status, starting with the following three subsystems makes sense, given these are the areas with the highest rate of FDA citations:
- Product and process controls
- Corrective and preventive actions
- Design controls
Production and process controls (P&PC)
P&PC received the most Form 483 observations out of all QS subsystems in FY2017. P&PC comprised 37 percent of total FY2017 Warning Letter (WL) citations. Additionally, 75 percent of Form 483 observations for P&PC were for domestic (United States) violations.
Observation Insights
Develops, conducts, controls, and monitors production processes to make sure a device aligns with specifications and establishes and maintains process controls to prevent deviations from occurring.
Section 820.50 accounted for the largest percentage of domestic P&PC Form 483 observations in FY2017. It deals heavily with ensuring that manufacturers are maintaining compliance across the supply chain by mandating that suppliers, contractors, and consultants are adhering to the same quality processes and standards. Plus, it requires detailed capture of purchasing data, including quality requirements and agreements from each entity.
Manufacturers that use hundreds of different suppliers and siloed documentation systems can struggle to understand if they have documented the processes, where such documentation is, and if it is up to date. More importantly, the critical question is if the teams have been trained on the processes and follow them. Inspections can happen with little or no warning and information may not exist in a way that can be easily recalled and shared with regulatory bodies.
Improvement points
- Shared specifications
- Collaborative evaluation
- Fast-track deviation process
To prepare for P&PC compliance, medical device manufacturers must establish well-documented, shared specifications that carry through to production, as well as an established collaborative evaluation and fast-track deviation processes. These measures provide a single, always-up-to-date view of the full product record across suppliers and siloed teams.
Arena QMS allows teams to control and access the full product design history and product record in one system. Audit procedures are much easier when all your quality actions and change processes are linked to the product record and are easily accessible and shareable. Integrating change management with design history files creates a controlled product record that improves traceability, reduces audit risk, and helps teams manage innovation without losing compliance or documentation integrity.
An integrated PLM and QMS platform makes it easier to maintain audit readiness by connecting quality records, product data, training, and document control. Read more about audit readiness with PLM and QMS.
Corrective and preventive actions (CAPA)
CAPA is critical for compliance because it connects the product, quality processes, and corrective actions from design through manufacturing. It’s also the second highest offender when it comes to FDA inspections. In FY2017, CAPA Form 483 observations constituted 33 percent of all observations and 32 percent of total WL citations.
Streamlining CAPA processes in QMS really helps make things run smoother by cutting down response times and making sure issues get resolved quicker and more effectively. Automating CAPA workflows helps organizations find root causes, put preventive measures in place, and reduce recurring problems. This supports regulatory compliance and encourages a culture of ongoing improvement. Improving CAPA processes in a QMS helps make products better and safer while lowering risks and boosting overall operational strength.
Observation insights
The majority of FY2017 QS WL citations and domestic CAPA Form 483 citations concern CAPA Section 820.100(a). This section requires manufacturers to have established procedures and documentation for taking corrective and preventive action that is appropriate for the type and importance of medical device being manufactured, including protocols for investigating the cause of nonconformities and communicating the findings to appropriate personnel and regulatory bodies.
Identifying the cause of the nonconformity can be difficult if there is not a connected view of the product and quality processes that quality managers, engineering, and operations can access. Furthermore, disconnected systems introduce risks that include incomplete application of actions across product lines or inconsistent and missed application of actions by teams, including the supply chain.
Improvement points
- Traceable CAPA loop
- Connected quality records
- Live updates
A critical must-have for CAPA compliance is a closed-loop system that’s easily traceable from issue to quality processes to product records. This system allows medical device companies to bring about better traceability and stronger controls by linking quality records with the complete bill of material (BOM), ensuring actions can be easily extended across related product lines when required. Well-defined quality and compliance processes help manufacturers connect issues with affected products, required actions, engineering changes, and supporting documentation.
Arena maintains all these relationships via live links, so users can instantly move from documents, parts, and multilevel assemblies to quality issues and actions or vice versa. Arena provides visibility between the CAPA records and related change orders, allowing internal auditors, quality and regulatory affairs, and other affected groups to instantly access and view all the issues, actions, and steps through final resolution in one central system.
Design controls (DES)
Many manufacturers see design validation as little more than an obstacle to getting to market. It is often rushed by manufacturers aiming to beat other products in competitive spaces. It can also be complex, as validation requires inputs from multiple teams across engineering, design, and quality management. In FY2017, the FDA recorded 455 DES Form 483 observations, accounting for 13 percent of all observations. Similarly, DES WL citations constituted 13 percent of all warning letter citations in the same year.
Observation insights
Section 820.30(g) requires manufacturers to have established and documented design validation processes, with an emphasized focus on risk management and testing the product in actual and simulated environments. The results should then be documented in the design history file (DHF). The DHF stays open and should be properly updated with product revisions as additional design validation occurs.
Improvement points
- Connected DHF and DMR
- Relational BOM
- Single platform
Complying with the DES subsystem requirements calls for a QMS that supports design control with seamlessly connected teams, products, and quality records. To demonstrate compliance and proper design controls, companies must be able to effectively manage the DHF and device master record (DMR) in context with each other and the entire product record. To manage the entire product record, medical device companies must have the ability to manage a complete BOM for electrical, mechanical, and software parts.
Arena QMS ensures the entire product record (e.g., parts, BOMs, AML, drawings, packaging, specifications) and quality record (e.g., CAPAs, DHFs, DMRs, NCMRs) are connected throughout the complete product lifecycle. This connected QMS methodology enables full traceability and creates stronger design controls that are aligned with FDA requirements. Integrating design control with document control ensures product data, requirements, and approvals remain accurate and traceable, helping manufacturers reduce risk while supporting faster development and audit readiness.
Enhancing Quality Subsystems to Avoid FDA Citations and Improve Compliance
Addressing the health of each quality subsystem is a characteristic of top performing medical device companies. Recent FDA data suggests that added attention is warranted on P&PC, CAPA, and design controls, and focusing on these frequently cited subsystems can reduce the likelihood of manufacturers receiving a Form 483, WL citation, or worse.
Technological developments, sustainability priorities, and changing market dynamics have led to a significant evolution of quality management systems for product development. These systems now incorporate trends such as advanced analytics, artificial intelligence (AI), cloud-based QMS, agile manufacturing, sustainable practices, customer feedback, and regulatory compliance.
Product-centric QMS solutions like those from Arena help tackle the biggest challenges by connecting the quality system to the product record, promoting communication and collaboration across teams, and helping quality leaders accelerate audits to a successful close.
Environmental Management Systems integrated with QMS systems significantly enhance sustainable product development, addressing rising global environmental challenges through improved compliance and innovative technologies. To learn more about how applying product-centric QMS helps your quality and product teams better meet FDA requirements, download our “7 Principles of Product-Centric Quality Management” ebook.
________________________________________________
Resources:
Overview of the Quality System Regulation for Medical Devices
FY2017 Annual FDA Medical Device Quality System Data