The 5 Essential Elements of a CAPA System
Introduction to CAPA Software
Medical device companies must have a compliant Corrective Action Preventive Action (CAPA) process in place to comply with FDA 21 CFR 820.100. A collaborative quality system lets you check this FDA requirement “box” while facilitating corporate objectives. In fact, even non-regulated companies in competitive industries require effective quality systems in order to compete in time to market, customer satisfaction, and cost control.
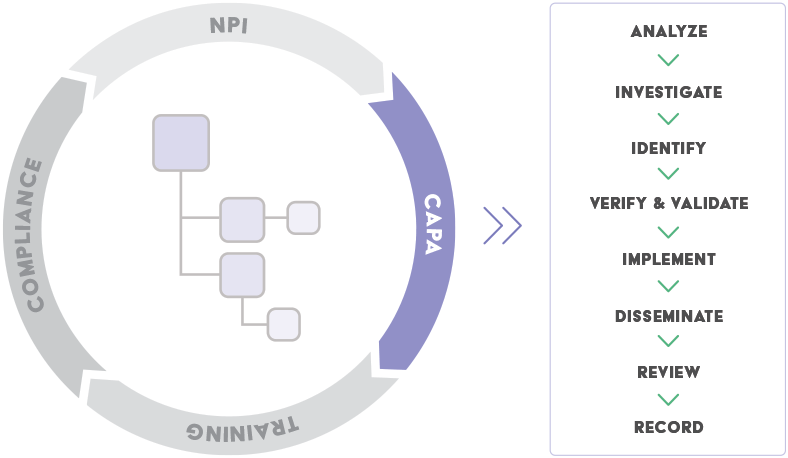
In order for the CAPA subsystem of a quality system to be FDA Compliant – here is what you must demonstrate:
“The purpose of the corrective and preventive action subsystem is to collect information, analyze information, identify and investigate product and quality problems, and take appropriate and effective corrective and/or preventive action to prevent their recurrence. Verifying or validating corrective and preventive actions, communicating corrective and preventive action activities to responsible people, providing relevant information for management review, and documenting these activities are essential in dealing effectively with product and quality problems, preventing their recurrence, and preventing or minimizing device failures. One of the most important quality system elements is the corrective and preventive action subsystem.”
FDA CAPA Inspection Guide
Home-grown, paper-based, or commercially available electronic systems may meet the minimum FDA and ISO 13485 requirements. However, most companies today want to go further than just checking the “box”, they want to capture and expand market share. If true, consider the following two questions:
- What are the basic elements of a compliant CAPA subsystem?
- What makes a Quality Management System (QMS) system, including the CAPA subsystem collaborative?
 CAPA Basics: Meet the Requirements
CAPA Basics: Meet the Requirements
Your CAPA processes will be managed by the CAPA subsystem of your quality management system. Your QMS may be a home-grown system or use a commercial quality management software solution. Either way, your system must adhere to the requirements listed in FDA 21 CFR 820.100. This is a significant hurdle for conducting business for FDA-regulated companies. Not everyone can overcome this hurdle.
According to the latest available statistics at the time of this publication1, the FDA issued 1441 findings referencing 21 CFR 820 (issued via Form 483) to medical device companies. An astonishing 197 of those observations, pertained to 820.100 or CAPA systems. Here are CAPA-related 483s by subcategory.
Number |
Description |
||
|---|---|---|---|
| 3130 21 CFR 820.100(a) | Lack of or inadequate procedures | Procedures for corrective and preventive action have not been [adequately] established. | 165 |
| 3696 21 CFR 820.100(b) | Documentation | Corrective and preventive action activities and/or results have not been [adequately] documented. | 32 |
2020 – FDA EIR database
A modern quality management system, with CAPA software built-in, helps avoid CAPA-related 483s in two ways. During internal audits, it provides visibility into issues before the inspector arrives. During FDA audits, it also simplifies demonstrating compliance with these regulations.
To meet FDA regulation 820.100, the CAPA subsystem of management processes of the QMS system must have:
- The ability to capture, review, approve, control, and retrieve established CAPA processes.
- The ability to capture and retrieve CAPA activities and/or results.
- A closed-loop process (including workflow and signoff) to facilitate verification or validation that the action is effective and does not adversely affect the finished device.
- A closed-loop process (including workflow and signoff) to ensure documentation of corrective and preventive action activities are established, defined, documented, complete, and implemented.
- The ability to capture, review, approve, control, and retrieve a corrective and preventive action procedure to determine when verification can be conducted in lieu of validation.
In summary, a QMS system must have the ability to capture, review, approve, control, and retrieve closed-loop processes.
The Difference: The Collaborative Quality System
 You didn’t start your company just so that you could keep the doors open with compliant systems. You want as many people as possible to benefit from your product. In business terms: you want to capture and expand market share. To do that, you need to accelerate time to market, maximize quality, and control costs. How do companies meet these goals? Collaboration.
You didn’t start your company just so that you could keep the doors open with compliant systems. You want as many people as possible to benefit from your product. In business terms: you want to capture and expand market share. To do that, you need to accelerate time to market, maximize quality, and control costs. How do companies meet these goals? Collaboration.
Collaborating among departments and throughout your supply chain is the key to delivering a high-quality product to market as quickly as possible. Collaborate throughout the product lifecycle–from the product conception to limited production to full production. Marketing, quality, engineering, field service, operations, suppliers, can all help bring the best product to market. It’s possible with the right system in place.
CAPA Software: 5 Essential Elements
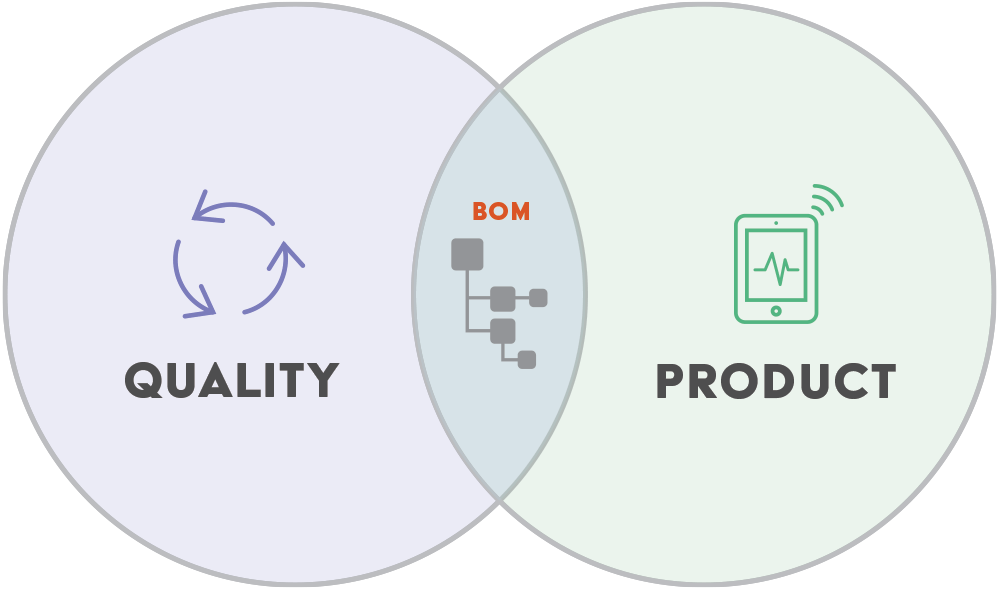
1. Organizes all documents and processes around the product bill of materials (BOM)
If you want all internal product teams and external supply chain partners to collaborate throughout the entire product lifecycle, you must manage and control 100% of your product data in a single, secure system. That system should contain all of your product data; electrical, mechanical, software, and documentation that is used to manufacture your products. This product-centric quality management system approach provides better visibility and traceability.
2. Supports the global enterprise
In order to include employees, suppliers, and customers in your processes, the system must easily support the extended enterprise which often spans the globe. Doing this in the first phase of implementation will have minimal impact on your IT resources and enable you to enjoy the benefits of collaboration more quickly and with less overhead.
3. Manages the CAPA process, including tasks, assignments, deadlines, all connected to affected products and documents
The collaborative quality system will enforce your CAPA business processes. With a process that can impact the entire organization, you need a system that can track key activities and provide a historical audit trail. By linking to the affected documents, users of the documents can see there is a pending action against the document and even open that action to come up to speed.
To speed implementation, the quality system should come configured with standard quality processes, like Eight Disciplines of Problem Solving (or “8D”).
Why reinvent the wheel? The best quality management systems (and the people who define, create, and support CAPA processes) understand current, established best practices. Having a versatile system to support proven best practice quality processes reduces the time to implement and yields key benefits as companies address quality issues before, during, and after shipping their products to market. The system should also be flexible enough to support custom processes when needed.
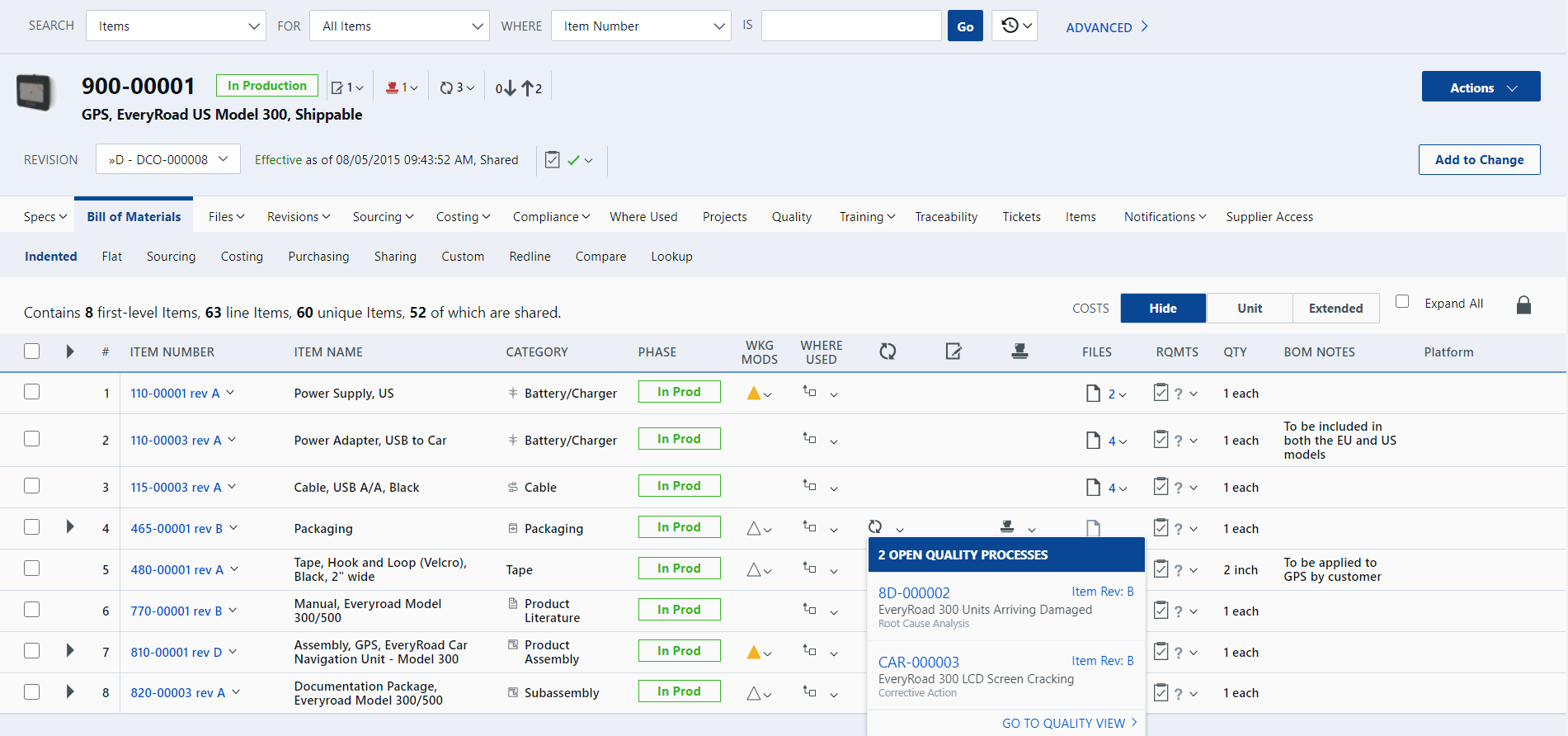
View of BOM showing pending quality issues for selected components
4. Integrates quality processes with the engineering change process
 A CAPA incident (or Corrective Action Request) may require an ECO to resolve a product problem. When that happens, you want the quality team to have visibility into the engineering change management process. Likewise, engineering teams should have visibility into quality issues that link directly to the affected product.
A CAPA incident (or Corrective Action Request) may require an ECO to resolve a product problem. When that happens, you want the quality team to have visibility into the engineering change management process. Likewise, engineering teams should have visibility into quality issues that link directly to the affected product.
Users should have visibility into all pending or released quality issues and engineering changes to enable easy research of all historical activity.
Integrating the engineering and quality processes eliminates the need for duplicate data entry or the need for cross-referencing with attribute data. You can quickly view and drill into related records.
5. Provides analytics for understanding the strengths and bottlenecks of your processes
With analytics, process owners see process metrics in real-time, so they can apply resources to meet business objectives. Modern analytics are so easy to use, you don’t need to program reports. You always have instant access to real-time data.
Conclusion
Using a compliant, product-centric Quality Management System, with connected CAPA capabilities helps you avoid CAPA-related FDA 483s. You will be able to maintain compliance with less anxiety, risk, and overhead.
It’s critical to include all of your product teams and suppliers as part of your CAPA process will shorten time to market, control spend, and improve product quality. This will enable you to speed time to resolution and ultimately expand market share by enhancing quality as you deliver safe and effective devices to your customers.
What Arena Does Better Than Document-Centric QMS
|
Product-Centric QMS Benefits
|
Functionality
|
|---|---|
|
Provides Enterprise-Wide Visibility |
|
|
Unifies Quality and CAPA Process With the Product Record |
|
|
Facilitates Quality Considerations During the Product Development Cycle |
|
|
Simplifies Enterprise Software System Validation |
|
|
Reduces Internal and External Audit Time |
|