5 Key Steps for Certifying Medical Devices in the Australian Market
Inside This Article

Today, many MedTech companies are setting their sights on Australia for expansion. The Australian medical device market is experiencing rapid growth, driven by increased healthcare spending, an aging population, and a strong MedTech workforce. Moreover, recent regulatory harmonization reforms in Australia offer an efficient pathway for manufacturers holding certifications from other regions.
Comparable overseas regulatory authorities include:
- Notified bodies under the EU MDR and EU IVDR
- U.S. Food and Drug Administration (FDA)
- Medical Device Single Audit Program (MDSAP)
- Health Canada
- Japan’s Ministry of Health, Labour and Welfare (MHLW), Pharmaceutical and Medical Devices Agency (PMDA)
- Singapore’s Health Sciences Authority (HSA)
Whether manufacturers are leveraging an existing certification or starting from scratch, they must have a thorough understanding of Australia’s Medical Devices Regulations to achieve compliance.
Here, we cover key steps for obtaining medical device certification in Australia and explore how a cloud quality management system (QMS) can help.
NAVIGATING AUSTRALIA’S REGULATORY LANDSCAPE
The Therapeutic Goods Administration (TGA) oversees the supply of medical devices and in vitro diagnostic (IVD) devices in Australia. Its primary role is to safeguard patient safety and foster innovation by establishing clear guidelines for device approval and monitoring.
Detailed compliance requirements, including device classification, conformity assessment procedures, and criteria for device authorization, are outlined in the Therapeutic Goods (Medical Devices) Regulations.1 Additional guidance is available in the Australian Regulatory Guidelines for Medical Devices (ARGMD).2
TGA’s Essential Principles
Companies must maintain clinical, design, and manufacturing evidence demonstrating their devices comply with TGA’s Essential Principles throughout the entire lifecycle.3
The following six Principles provide a framework for ensuring devices meet necessary performance and safety standards before market release:
- Medical devices ensure patient health and safety
- Design and production methods conform with safety principles
- Medical devices perform according to their intended use
- Devices are designed and produced to ensure long-term safety
- Transport and storage methods do not adversely affect device characteristics and performance
- Medical device benefits outweigh any undesirable effects
Medical Device Certification and Inclusion in the Australian Register of Therapeutic Goods (ARTG)
Medical and in vitro diagnostic devices require a conformity assessment certification and listing in the ARTG to be lawfully sold in Australia. As part of the conformity assessment, the TGA evaluates clinical data, design records, quality processes, and technical documentation to determine if the device performs as intended and adheres to the Essential Principles.
Upon receiving a certificate, companies can apply for their device to be listed in the ARTG. This searchable online database is accessible to the public and provides the following product information:
- Product name and summary
- License details (device classification, etc.)
- Manufacturer and sponsor details
- ARTG identifier number
- Start date (date the product was added to the ARTG)
- Effective date (last date a change was made to a product entry)
STEPS FOR OBTAINING TGA MEDICAL DEVICE CERTIFICATION AND APPROVAL
Once you have a firm grasp of TGA’s regulatory framework, you can begin strategizing and executing the required actions to ensure compliance. Below are five crucial steps to certify your device and secure TGA approval.
1. Classify your device.
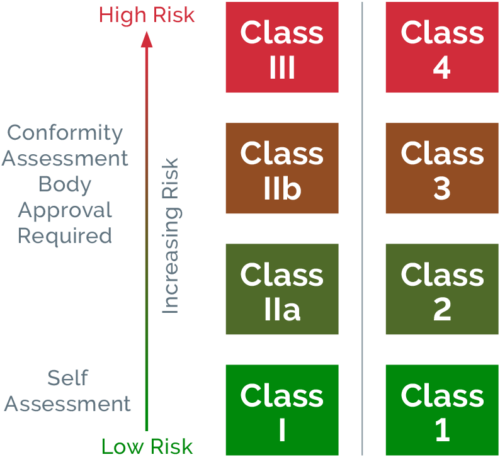
Refer to the classification rules that are outlined in Schedules 2 and 2A of the Therapeutic Goods (Medical Devices) Regulations to accurately classify your device. Australia’s medical device classification system mirrors that of the EU, categorizing devices according to their intended use and risk they pose to patients.
Medical Device
|
Risk |
Examples |
| Class I | Low |
|
|
Class Is (Supplied Sterile) |
Low-Medium |
|
|
Class Im (Incorporating a Measuring Function) |
|
|
| Class IIa |
|
|
| Class IIb | Medium–High |
|
| Class III | High |
|
| AIMD (Active Implantable Medical Devices) | High |
|
IVD devices are classified according to their intended use and the public health or personal risk that may arise from an incorrect result.
IVD Device
|
Risk |
Examples |
|
Class 1 |
No public health risk Low personal risk |
|
|
Class 2 |
Low public health risk Moderate personal risk |
|
|
Class 3 |
Moderate public health risk High personal risk |
|
|
Class 4 |
High public health risk |
|
2. Designate an Australian sponsor.
Appoint a local sponsor to serve as a liaison between the TGA and your organization. This representative will submit a conformity assessment application as well as a medical device application to be included in the ARTG. They will also manage post-market surveillance activities like adverse event reporting.
3. Compile QMS documentation.
Update your QMS records (including the technical file) to align with TGA’s Essential Principles and comply with ISO 13485. Key documentation includes:
- Quality policy and manual
- Management team responsibilities
- Resource management (i.e., infrastructure, equipment, and supply chain)
- Detailed product specifications and guidance on intended use
- Plan and processes for ensuring document control and protection of sensitive information
- Product realization—planning, design, development, production, and service provision
- Risk management plan
- Processes for post-market surveillance
- Clinical data
4. Obtain a TGA-issued conformity assessment certificate.
Submit a conformity assessment application and supporting evidence via the TGA Business Services Portal. Your device classification will determine the type of evidence that is required and extent of the evaluation. For low-risk devices (Class I), manufacturers generally perform a self-assessment and provide a declaration of conformity. In contrast, higher-risk devices (Class IIb, III, or Class 4 IVD) require an extensive review of the manufacturer’s QMS, clinical evaluations, and technical documentation. In this instance, an assessor may conduct the review via an on-site or off-site audit.
The review process lasts approximately 160 to 225 business days.4 If you already have a certification from a comparable overseas authority (e.g., CE Mark), you can use TGA’s abridged assessment route, which reduces the review timeline.
5. Apply for a device listing in the ARTG.
Submit a medical device application via the TGA Business Services Portal. Once approved, the TGA will issue your device’s ARTG listing number and certificate.
POST-MARKET SURVEILLANCE AND VIGILENCE
Registering devices with the ARTG is just the beginning. Subsequently, companies are responsible for monitoring their devices to ensure compliance with TGA’s Essential Principles. This includes reporting of adverse events and submission of annual safety and performance reports. In turn, the TGA will investigate adverse events and conduct periodic reviews of the manufacturer’s QMS and technical file.
STREAMLINE TGA COMPLIANCE WITH A PRODUCT-CENTRIC QMS
Navigating TGA’s conformity assessment can be overwhelming without the right resources and technology. Arena QMS alleviates this burden by centralizing all your product and quality records in a single cloud platform. By managing linked relationships between design files, bills of materials (BOMs), supplier information, technical documentation, and any changes or quality issues, Arena offers your teams greater control, traceability, and visibility. This product-centric approach helps mitigate compliance risks as you work toward certification.
Arena’s quality templates enable you to quickly build post-market surveillance reports and other essential documentation to suit your needs. Additionally, everything is kept up to date and easily retrievable to expedite audits.
For additional insights on navigating Australia’s medical devices regulations, explore our ebook.
References